Do I have a liver problem?
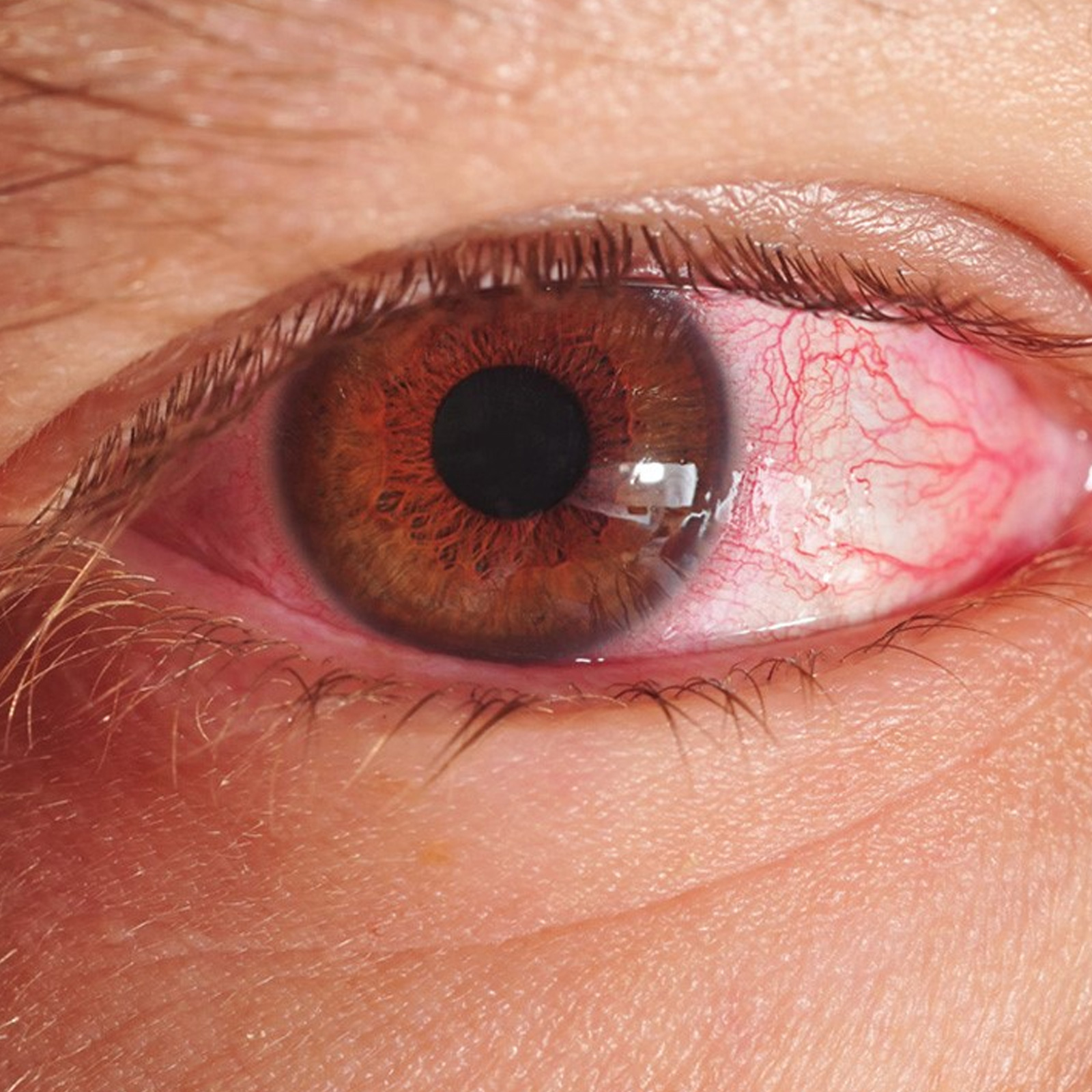
Q: I was suspected to have Jaundice. The tests of bilirubin showed 3.4 (conj. 0.9 & unconj. 2.5) and 2.2 (conj. 0.8 and unconj. 1.4) and SGPT as 36 and 34. The test for HBsAg was negative (using ELISA method). The ultrasonography of the whole abdomen revealed that only the size of the liver is 139 mm (co-axial length) and spleen is 136 mm (co-axial length), while the rest are fine. The test for Liver Function revealed that everything was fine except the Bilirubin level shows 5.7 (conj. 0.9 & unconj. 4.8) and SGPT as 36. I do not have any vomiting tendency nor do I feel weak. I have a sound sleep, my urine colour is not yellowish, but my eye ball is having some yellowish colour. Sometimes there is formation of gas in the stomach and the stool is not upto the mark. I drink a lot of water (around 4 & 1/2 ltrs.) daily. The doctor has said that this is a harmless jaundice and is due to some enzyme deficiency which is by birth. He also has suggested to lead a normal life and said there is nothing to worry. Now may I please be suggested that if this has really nothing to worry or if there is any medication available to rectify the problem. Looking forward for your valuable reply.
A:I think your case just needs to be followed. Dubin-Johnson syndrome (DJS) is a type of hereditary hyperbilirubinaemia that was first described independently in 1954 by Dubin and Johnson and by Sprinz and Nelson. Hereditary hyperbilirubinaemias can be divided into conjugated and unconjugated forms. While Gilbert and Crigler-Najjar syndromes are examples of the unconjugated hyperbilirubinaemias, Dubin-Johnson and Rotor syndromes represent the 2 types of familial conjugated hyperbilirubinaemias. Both types of conjugated hyperbilirubinaemias have a relatively benign course, but it is important to establish the diagnosis to spare patients from undergoing multiple unnecessary procedures and to exclude other more serious causes of hyperbilirubinaemia. Pathophysiology: DJS is an autosomal recessive disorder that is caused by a mutation in the gene responsible for the human canalicular multispecific organic anion transporter (cMOAT) protein. This protein mediates ATP-dependent transport of certain organic anions across the canalicular membrane of the hepatocyte. A defect in the cMOAT protein results in impaired hepatobiliary transport of non–bile salt organic anions and is thought to be responsible for the conjugated hyperbilirubinaemia and for the accumulation of hepatocellular pigment. Age: Patients with DJS tend to develop nonpruritic jaundice during their teen years. Although, most patients are asymptomatic, some complain of nonspecific right upper quadrant pain, which has been attributed to the anxiety associated with prolonged diagnostic testing. Subclinical cases can become evident during pregnancy or following the initiation of oral contraceptives. A thorough family history can reveal a history of jaundice in an autosomal recessive pattern. Aside from the presence of jaundice, physical examination findings are generally normal, with the exception of possible hepatosplenomegaly. Hyperbilirubinaemia and clinical icterus can be worsened by intercurrent illnesses, by drugs that can decrease hepatic excretion of organic anions (eg, oral contraceptives), and by pregnancy. Causes: DJS is an autosomal recessive disorder that is caused by a mutation in the gene responsible for the cMOAT protein. A liver biopsy is not necessary for diagnosis. Patients may be noted to have a dark liver during routine surgeries (eg, cholecystectomy), prompting biopsy.